An Analytical Thesis on Machine-learning Modeling of Chemically Disordered Metallic Alloys
Table of Contents
- Analytics in metals modeling
- Contrast with brute force data generation
- Cause and effect in training data diversity
- From lab to industry and future tooling
Materials used in aerospace, energy, and computing push performance beyond conventional boundaries. Yet predicting how a new metal will behave inside a rocket or on a chip is hindered by chemical disorder in most solid materials. Traditional simulations struggle to represent every local atomic arrangement accurately, inflating cost and delaying innovation. MIT researchers propose a path forward: machine-learning models trained on information theoretic training datasets designed to capture the diversity of atomic environments in disordered metals.
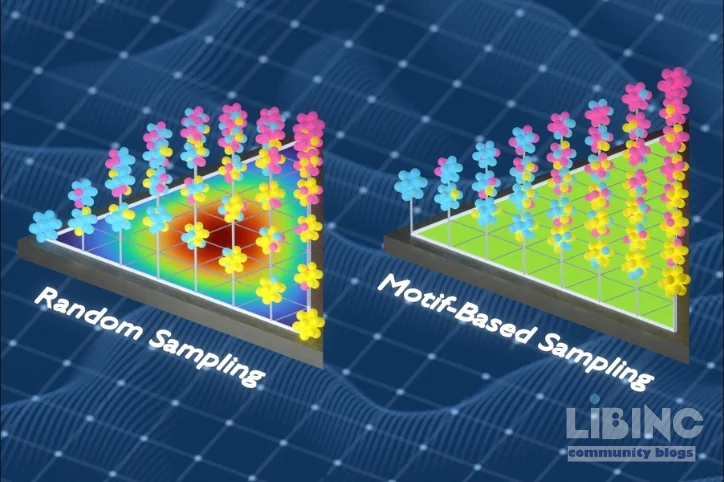
The core idea is to swap atoms in samples to expose the model to unusual local environments, reducing redundancy and increasing information content. This information theoretic approach builds training datasets that reflect the true complexity of solid metals, unlike brute force sampling that repeats familiar motifs. When trained on these datasets, the models predict properties with greater fidelity across a range of compositions and temperatures, including phase diagram behavior. The implication is explicit: you can design and process alloys with predictive confidence rather than rely on trial and error experiments. This is crucial as industry seeks sustainable steels, aerospace alloys, and components that tolerate extreme conditions.
The study demonstrates that a data driven path can translate chemistry into reliable physical predictions. The approach targets chemically disordered phases where local environments vary widely, making standard models brittle. The MIT team shows that a carefully curated training set improves transferability, allowing the same framework to handle diverse alloys without rebuilding the model for every new composition. The research emphasizes that a reduction in data excess does not come at the cost of accuracy; instead it unlocks better generalization by focusing learning on meaningful diversity. The result points toward a practical shift in how materials are designed, processed, and validated, especially when experiments are expensive or hazardous.
Finally, the authors discuss how the method can extend beyond metals to other classes of materials such as semiconductors. The promise lies in a framework that aligns with how engineers think about phase stability and performance under real processing conditions. In short, machine learning for disordered materials becomes a tool that complements traditional experiments and atomistic simulations, enabling designers to navigate complex landscapes with fewer expensive trials and more informed decisions.
Analytics: From order to disorder to prediction
Within the analytics frame, the key challenge is building a predictive model that respects the chemistry of disordered metals. Atomic interactions in a disordered lattice depend on the immediate neighborhood, which can vary from site to site in ways that standard, orderly datasets fail to capture. The breakthrough comes from constructing training datasets that intentionally maximize informational diversity. This means exposing the model to rare and intermediate local environments that may influence phase formation, ordering tendencies, and mechanical behavior in surprising ways.
The work proceeds by first quantifying chemical complexity using an information theoretic lens. The local arrangement of atoms is not just a count of species; it is the spatial pattern and the distances between neighbors that govern bonding and energetics. By measuring the frequency and spacing of substructures within a sample, researchers can assign a complexity score to a given alloy configuration. This score guides the sampling strategy so that the training set contains a wide array of unique environments rather than many repetitions of the same motif. Such diversity is essential for enabling the model to interpolate or extrapolate to compositions and temperatures it has not yet seen.
Next, the team implements a dynamic data augmentation protocol. They swap out atoms in representative configurations to reduce redundancy while preserving physical realism. This process uncovers local environments that would otherwise be underrepresented in a random sampling scheme. The result is a training corpus that is not merely large but informative, where each example contributes new information about how a disordered alloy behaves under typical operating conditions. The shift in data philosophy is as important as the scale of data, because it changes what the learning algorithm can learn about chemical bonding and local energetics.
With these datasets in hand, a family of machine learning models is trained to map local atomic environments to properties of interest. The models learn to relate energy, bonding patterns, and local order to macroscopic outputs such as phase stability, phase boundaries, and mechanical response. Importantly, the predictive task is not limited to a single material. The same approach demonstrates robustness across a group of chemically diverse alloys, suggesting that the underlying representation captures general features of disordered bonding rather than material specific idiosyncrasies. This generality is what makes the method attractive for industry, where a single workflow could inform multiple design choices across products and applications.
From a methodological standpoint the core advance is that the training set becomes a learning signal itself. Rather than passively accumulating data, the dataset actively teaches the model where the chemistry matters most. The learning objective aligns with physical reasoning: to predict material properties with high fidelity requires a faithful representation of local chemistry and its variations under different thermodynamic conditions. The consequences go beyond accuracy. When models encapsulate realistic chemical diversity, they are more reliable for decision making in environments where experimental data are scarce or expensive to obtain.
Figure and table outcomes in the study illustrate that models trained on these information rich datasets outperform larger models that rely on standard sampling strategies. The comparison highlights a crucial point: computational efficiency is not solely about fewer parameters. It is about smarter data that reveals the nuanced interplay between local structure and bulk properties. The practical implication is that predictive capabilities can scale with real world complexity without a proportional increase in compute time or data volume. This directly supports faster iteration in alloy design and processing optimization.
Contrast: why brute force data generation fails and the alternative
Traditional materials modeling has often leaned on brute force data generation. In the past, researchers would generate extensive datasets by exhaustively sampling configurations and running atomistic simulations to convergence. While this brute force approach can yield accurate snapshots for a single composition, it quickly becomes impractical as the number of variables grows. For chemically disordered alloys, the combinatorial space of local environments expands rapidly with composition, temperature, and pressure. The result is a dataset that is either prohibitively large or insufficiently representative of rare but influential environments.
As a consequence, models trained on brute force data exhibit limited transferability. They capture the behavior of a narrow slice of the composition space and then stumble when asked to interpolate beyond that slice. In practice this means poor predictions for phase diagrams outside the original training zone, mischaracterized ordering tendencies, and unreliable estimates of mechanical properties under harsh conditions. The study argues that the fundamental reason is not merely data volume but data quality: repetitive examples reinforce known patterns while rare local motifs that govern phase transitions remain underrepresented. The end result is a model that looks confident but fails when applied to novel alloys or processing routes.
The information theoretic sampling strategy changes this calculus. By actively seeking diverse local environments and pruning redundant examples, the training dataset becomes a compact yet richly informative resource. The method does not eliminate brute force; it refines the input to what the learning algorithm must learn. The payoff is twofold. First, predictive accuracy improves across a broader spectrum of compositions and conditions. Second, the required compute and data generation time shrink relative to a conventional brute force campaign. The researchers show that smaller, smarter data can outperform larger, unimodal datasets when the latter are not sufficiently diverse.
In addition to accuracy, the approach emphasizes relevance to real processing decisions. Phase diagrams emerge as a central tool for materials design because they summarize stability across temperature and composition. The authors demonstrate that phase boundaries predicted by their models align closely with experimental measurements, a validation that strengthens the case for deploying these models in everyday design tasks. This alignment is not incidental; it reflects the models' ability to capture subtle energetic biases toward certain local chemical configurations that drive phase formation and transformation. The result is a practical bridge from computational prediction to processing choices such as welding, casting, and heat treatment.
From the standpoint of industry practice, the key contrast is not just accuracy but reliability under processing variability. Brute force approaches can falter when real world conditions push dynamics beyond the training envelope. The information driven training strategy targets those edges explicitly, equipping engineers with more trustworthy predictions as they explore new alloy compositions and novel processing windows. The method thereby supports risk management in materials development, reducing the likelihood of costly mispredictions during scale up or field deployment.
The discussion also touches on broader implications for the field of computational materials science. The capacity to generate informative datasets at scale suggests a new paradigm where data strategy is as important as algorithm design. It invites a shift from a culture of dataset size to a culture of dataset quality and representativeness. In this sense the work moves the discipline toward data driven discovery that preserves physical interpretability while embracing the complexity of real world materials systems.
The next step is to extend the approach to more materials classes and to integrate it with workflows that materials engineers already rely on. The aim is to embed the training procedure into product development cycles so that predictions feed directly into design choices and processing protocols. The promise is not merely faster simulations, but better, more robust guidance for creating materials that perform under demanding service conditions and reduce life cycle costs while improving safety and reliability.
Cause and effect in training data diversity
The core argument is causal rather than purely correlational. When the training dataset highlights a broad spectrum of local chemical environments and energetic biases, the learning model learns how small changes in composition or temperature ripple through the phase landscape. This is a shift from pattern recognition to causal inference about material response. The consequence is more credible predictions of phase stability, ordering tendencies, and property changes as alloys are processed under real world conditions.
One practical signal of this causal improvement appears in phase diagram predictions. Phase diagrams summarize which phases are stable at given temperatures and compositions, a central tool for alloy design. The study shows that models trained on diverse, information rich data reproduce phase boundaries with a fidelity comparable to experiments under a variety of processing routes. This implies engineers can rely on the model to forecast how a new alloy will behave during welding or heat treatment, before committing time to costly experiments. In other words, the model provides a trustworthy compass for processing decisions in the materials design loop.
Beyond phase diagrams, the approach improves predictions of mechanical properties that respond to local ordering and microstructure. Elastic moduli, yield strength, and radiation tolerance, for example, are sensitive to how atoms cluster or distribute in the lattice. A model informed by diverse local environments can capture how small shifts in composition alter the balance between brittle and ductile responses. This capability is essential for designing materials that remain strong and damage tolerant in harsh environments, such as space or nuclear settings. The causal insight here is clear: diversity in training data translates into robust, transferable predictions with practical design value.
Another facet concerns transferability across compositions. A model trained with a carefully curated dataset should generalize beyond the exact alloys in the training set. The research shows promising signs of this transferability, suggesting that a single modeling framework could cover a family of alloys rather than require bespoke models for each material. That generalization is the practical payoff for industry, which seeks scalable workflows rather than bespoke, one off solutions for every new material. In this sense the data strategy acts as a catalyst for broader exploration of material spaces while maintaining reliability.
From a theoretical perspective, the work invites deeper questions about the nature of disorder and energetics in metals. The subtle energetic biases toward particular local configurations reveal that disorder is not purely random but organized by thermodynamics in ways that can be learned by neural networks when trained on adequately diverse data. This reframing invites a tighter integration between machine learning and physical chemistry, where statistical patterns in local chemistry reflect fundamental energetic landscapes. The result is a more nuanced picture of how disordered alloys assemble into phases under real world processing conditions.
To translate these insights into practice, researchers are pursuing easier integration with existing tools used by materials engineers. They are exploring streamlined interfaces and workflows that let designers query phase stability and property predictions within familiar design environments. The end goal is a user friendly, industry ready package that preserves the rigorous physics underpinning the models while fitting into the cadence of product development and manufacturing planning. The emphasis is on utility as much as on accuracy, with the ultimate aim of accelerating the discovery of materials that meet stringent performance and sustainability targets.
From lab to industry and future tooling
Industry uptake hinges on aligning new modeling capabilities with existing workflows and decision points in manufacturing. The approach described here emphasizes practical integration: the training data strategy, model architectures, and validation against experiments are presented with an eye toward plug and play compatibility. This means leveraging familiar tools for materials design, such as phase diagram analysis, alloy design strategies, and standard workflows for heat treatment and welding. The objective is to lower barriers so engineers can incorporate machine learning into daily practice rather than treating it as a separate research project.
The paper also addresses validation. By comparing predictions to experimental measurements of atomic ordering and phase stability, the authors build confidence that the models capture not only abstract patterns but concrete material behavior. This validation is critical for industrial adoption, as it demonstrates that the approach can connect molecular scale interactions with macroscopic performance. It also helps quantify the risk of relying on simulations for process optimization, offering a clearer path to cost reduction and faster time to market.
From the perspective of deployment, the authors discuss the need for user friendly interfaces and workflows that engineers already rely on. They highlight the importance of compatibility with data management practices, reproducibility of results, and transparent reporting of uncertainties. Such considerations matter because industrial teams want to trace predictions back to the underlying physics and validated data before adjusting processing parameters or selecting material grades. The practical upshot is a more predictable, auditable design process that can scale from single labs to multi site operations.
In parallel, the research lays groundwork for extending the method beyond metallic alloys. Semiconductors, ceramics, and composite materials exhibit their own forms of disorder and local environment dependence. A generalized information theoretic data strategy could enable similar improvements in predictive capability across material classes. The researchers anticipate adapters that map the same learning principles onto different chemistries and bonding networks, preserving the core idea that diversity in local environments drives predictive power. This cross domain potential positions the approach as a cornerstone of data driven materials discovery across industries.
Looking ahead, the work invites several lines of refinement. One involves automating the discovery of informative environments, further reducing human expertise required to curate datasets. Another focuses on uncertainty quantification, so engineers can gauge the confidence of model predictions under novel processing conditions. A third concerns integration with experimental design, enabling closed loops where simulations guide experiments, and experimental outcomes update the model in near real time. Each of these directions aims to sharpen the link between in silico predictions and real world performance, delivering a robust, scalable path from concept to certification.
In short, the combined thrust of this approach is to convert complex local chemistry into actionable material design intelligence. Through smarter data, the models become reliable partners in the materials design process rather than black boxes that require endless experiments to validate. The result is a more efficient, more resilient path from discovery to deployment, with broad implications for sustainable steel development, aerospace alloys, and advanced computing substrates. The research marks a meaningful step toward a future where data driven predictions inform every significant materials decision on the factory floor and in the design studio.
As a final reflection, the authors acknowledge support from the U S Air Force Office of Scientific Research, underscoring the strategic importance of durable, high performing materials in defense as well as civil applications. Yet the potential reach extends beyond national programs to any industry seeking to accelerate materials innovation without sacrificing reliability. The combination of chemically aware models and information theoretic data design represents a disciplined advance rather than a speculative concept. Its maturation will depend on continued validation, thoughtful integration, and community adoption across the materials engineering ecosystem.
In the end these developments do not promise instant miracles. They promise a more principled approach to materials design that respects the realities of disorder and the costs of experimentation. By recognizing that the diversity of atomic neighborhoods is not a nuisance but a source of information to be harnessed, engineers can steer alloy development with greater confidence, reduce costly dead ends, and deliver materials that perform as intended under demanding service conditions. That is the practical advantage and the strategic impetus behind machine learning for chemically disordered metallic alloys.
Overall, the trajectory suggested by this work points toward a future where data science and materials science co evolve. The goal is not a single algorithm but an ecosystem of training strategies, validation protocols, and industry ready tools that together make disordered materials easier to design, study, and deploy. If this vision is realized, the material choices that define our most demanding technologies will be informed by models that truly understand the subtle patterns of local chemistry and their macroscopic consequences.
To close, the central takeaway is clear. By focusing data acquisitions on informative diversity, machine learning can deliver reliable, transferable predictions for disordered metals. This means faster innovation cycles, cheaper experimentation, and more robust materials for the products that shape our future. The field moves from simply simulating atoms to guiding real world engineering decisions with a level of confidence once thought unattainable. That is the practical promise of machine learning for chemically disordered metallic alloys.
Uncertainty-aware predictions for disordered metals
To translate accuracy into reliability, practitioners need explicit confidence bounds for predictions across unobserved compositions and processing conditions. An uncertainty quantification (UQ) layer, built on ensemble learning and principled calibration, provides prediction intervals for phase stability and mechanical properties in chemically disordered alloys.
| Aspect | Brute Force Data | Info-Theoretic Sampling | Impact | Notes |
|---|---|---|---|---|
| Diversity | High repetition | Targeted, diverse motifs | Better generalization | Captures rare environments |
| Data Volume | Large | Smaller but richer | Data efficiency | Less waste |
| Transferability | Limited | Broad across compositions | Widely applicable | Industry friendly |
In practice, ensembles reveal when the model is extrapolating and help bound risk during design decisions, such as weldability windows or heat-treatment schedules. The next step is to couple UQ with monitoring against real experiments to close the loop.
Beyond reliability, UQ supports risk-aware design in industry by signaling when a prediction lies outside validated regimes, guiding experimental validation and reducing costly failures.
- Establish baseline models and data diversity
- Define target compositions and processing windows
- Build ensembles across different architectures
- Quantify and calibrate uncertainty
- Use hold-out sets and cross-validation
- Validate against known phase diagrams
- Integrate into design workflows
- Embed predictions in phase stability maps
- Link to processing parameter spaces
In aerospace and energy alloys, these practices translate into confident design margins and reduced trial-and-error rounds, accelerating safe deployment.
- Establish baseline models and data diversity
- Define target compositions and processing windows
- Build ensembles across different architectures
- Quantify and calibrate uncertainty
- Use hold-out sets and cross-validation
- Validate against known phase diagrams
- Integrate into design workflows
- Embed predictions in phase stability maps
- Link to processing parameter spaces
What is information theoretic data design in materials ML?
It is a strategy that prioritizes informative diversity in training data by selecting configurations that maximize information about local environments, rather than simply maximizing data volume. This approach helps models learn general rules governing disordered alloys and their properties.
How does uncertainty quantification improve predictions for disordered alloys?
UQ provides prediction intervals and calibrated confidence, indicating how much trust to place in outputs when encountering unseen compositions or processing conditions. It helps avoid overconfident extrapolations and guides risk-aware decisions.
How can these models be integrated into existing design workflows?
Predictions can feed phase diagrams, property estimates, and processing windows directly into design environments and data management systems, enabling engineers to compare alternatives with quantified risk.
What evidence supports transferability across alloy families?
Diversity-focused training yields models that generalize to related compositions, maintaining accuracy in phase boundaries and mechanical properties without retraining for every new alloy.
How can a company begin adopting this approach?
Start with a pilot dataset using informative sampling, train an ensemble with calibrated uncertainty, validate against experiments, and integrate predictions into existing design pipelines for gradual adoption.





Add a comment
To comment, you need to register and authorize
Comments